Let’s discuss the question: how to sort sam file. We summarize all relevant answers in section Q&A of website Achievetampabay.org in category: Blog Finance. See more related questions in the comments below.

Why do BAM files need to be sorted?
Indexing a genome sorted BAM file allows one to quickly extract alignments overlapping particular genomic regions. Moreover, indexing is required by genome viewers such as IGV so that the viewers can quickly display alignments in each genomic region to which you navigate.
How are BAM files sorted?
BAM files are sorted by reference coordinates (samtools sort) Sorted BAM files are indexed (samtools index) Sorted, indexed BAM files are filtered based on location, flags, mapping quality (samtools view with filtering options)
Understanding SAM/BAM file specifications
Images related to the topicUnderstanding SAM/BAM file specifications
Can you merge SAM files?
MergeSamFiles (Picard) Follow. Merges multiple SAM and/or BAM files into a single file. This tool is used for combining SAM and/or BAM files from different runs or read groups into a single file, similarl to the “merge” function of Samtools (http://www.htslib.org/doc/samtools.html).
Does samtools merge sort?
Merge multiple sorted alignment files, producing a single sorted output file that contains all the input records and maintains the existing sort order. The output file can be specified via -o as shown in the first synopsis.
What is BAM Sorting?
A bam is coordinate sorted if the reads are sorted by coordinates. So reads from the beginning of the first chromosome are first in the file. That’s what samtools sort is for. The opposite would be ‘sorted by name’ in which reads are sorted by their read ID.
How do I read a BAM file?
BAM files can be opened from remote locations (ftp, http) and from local computers. For viewing BAM files, an index file must be found in the same directory as the BAM file. The index should be named by appending “. bai” to the BAM file name.
What does SAMtools Flagstat do?
DESCRIPTION. Does a full pass through the input file to calculate and print statistics to stdout. Provides counts for each of 13 categories based primarily on bit flags in the FLAG field. Information on the meaning of the flags is given in the SAM specification document <https://samtools.github.io/hts-specs/SAMv1.pdf>.
How long should SAMtools sort take?
We compared the sorting speed of a 25Gb unsorted BAM file with SAMtools and sambamba. Our results show that sambamba was 2x faster than SAMtools. The following violin plot shows that SAMtools took 20 minutes while sambamba could sort the same file in 10 minutes.
How do I open a BAM file in Linux?
The way to visualise BAM files as text in Linux is with tools like samtools. If you want to see the raw binary information as slightly more human-readable, you can run hexdump -C <file.
How do you convert BAM to bigWig?
- Objective.
- Method 1: RPM track file from BAM file. Getting the number of mapped reads.
- Method 2: RPM track file from BAM file.
- View the Results in IGV.
- View the results at UCSC.
- Create a bigWig header line. Covid Track Server Countermeasure.
What does samtools collate do?
DESCRIPTION. Shuffles and groups reads together by their names. A faster alternative to a full query name sort, collate ensures that reads of the same name are grouped together in contiguous groups, but doesn’t make any guarantees about the order of read names between groups.
What does samtools view do?
Samtools is a set of utilities that manipulate alignments in the SAM (Sequence Alignment/Map), BAM, and CRAM formats. It converts between the formats, does sorting, merging and indexing, and can retrieve reads in any regions swiftly. Samtools is designed to work on a stream.
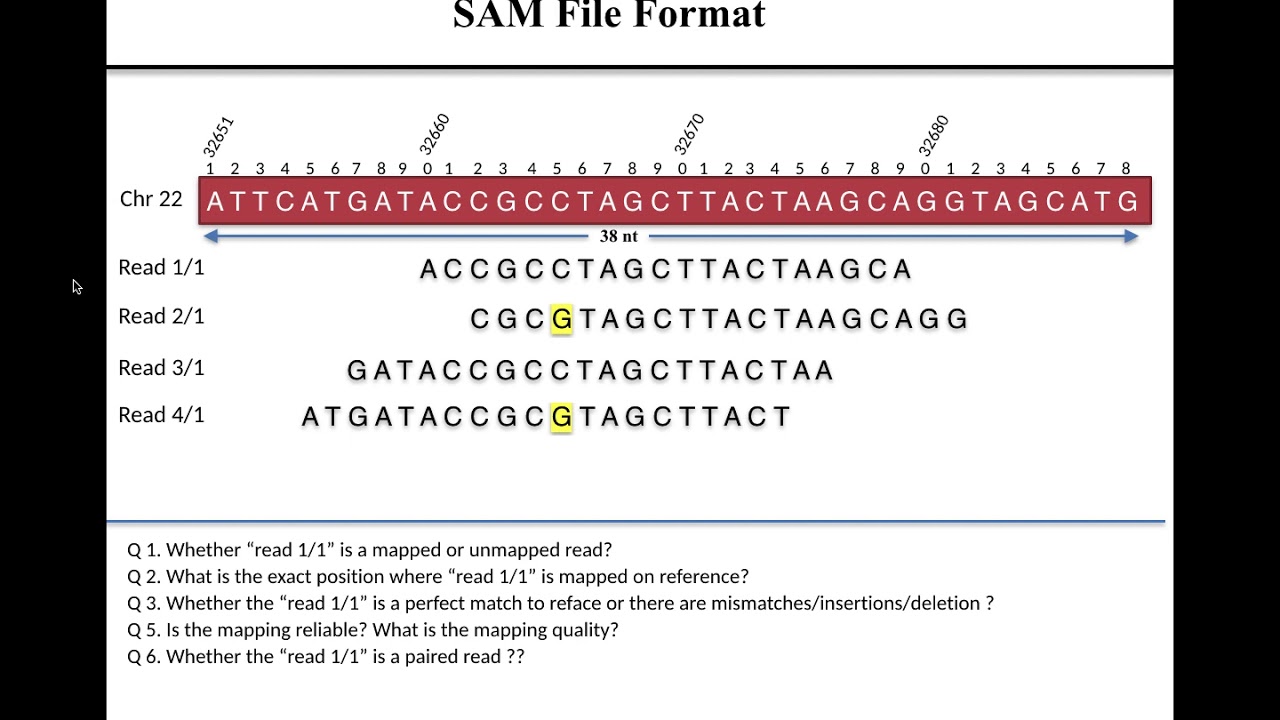
Lecture 1 SAM File Format
Images related to the topicLecture 1 SAM File Format
What does SAMtools Fixmate do?
samtools fixmate – fills in mate coordinates and insert size fields.
Can you merge bigwig files?
bigWigMerge : Merge together multiple bigWigs into a single bw file. The signal values are just added together to merge them.
How do you convert BAM to Fastq?
If your BAM alignments are from paired-end sequence data, one can use the -fq2 option to create two distinct FASTQ output files — one for end 1 and one for end 2. When using this option, it is required that the BAM file is sorted/grouped by the read name.
What are Sam tools?
SAMtools is a set of utilities for interacting with and post-processing short DNA sequence read alignments in the SAM (Sequence Alignment/Map), BAM (Binary Alignment/Map) and CRAM formats, written by Heng Li. These files are generated as output by short read aligners like BWA.
What is BAM file?
A BAM file (. bam) is the binary version of a SAM file. A SAM file (. sam) is a tab-delimited text file that contains sequence alignment data. These formats are described on the SAM Tools web site: http://samtools.github.io/hts-specs/.
What is SAMtools Mpileup?
The SAMtools mpileup utility provides a summary of the coverage of mapped reads on a reference sequence at a single base pair resolution. In addition, the output from mpileup can be piped to BCFtools to call genomic variants.
How do I convert a BAM file to a VCF file?
It’s not really possible to convert bam to vcf . bam is a mapping file, it does not contain the information about variants, this information needs to be inferred in process called variant calling. I find important to mention that it’s not just a different format of the same thing.
What is in a SAM file?
SAM files are a type of text file format that contains the alignment information of various sequences that are mapped against reference sequences. These files can also contain unmapped sequences. Since SAM files are a text file format, they are more readable by humans and will be used as the examples for this section.
What is a BAM index?
A bam file is a binary blob that stores all of your aligned sequence data. You can view what’s in the bam file using “samtools view bamfile. bam | less”. Bam files can also have a companion file, called an index file. This file has the same name, suffixed with .
What are properly paired reads?
Proper pairing means reads are in Read1 forward, Read2 reverse orientation or Read1 reverse, Read2 forward orientation. To extract single end reads from a bam file (-b) that map with mapQ≥30 including the bam file header (-h).
How to copy SAM file and SYSTEM file with CMD
Images related to the topicHow to copy SAM file and SYSTEM file with CMD

How do I extract unmapped files from a BAM file?
…
Filter Alignments for unmapped pairs
- An unmapped read whose mate is mapped.
- A mapped read who’s mate is unmapped.
- Both reads of the pair are unmapped.
What are secondary reads?
So it is clear that secondary reads are for cases where the same part of sequence aligns to multiple locations and supplementary reads are where (mostly) non-overlapping parts of a sequence align to multiple locations.
Related searches
- samtools sort unmapped
- samtools sort multiple bam files
- how to view bam file in linux
- samtools sort vcf
- Sort bam file
- Samtools index
- how to sort a folder by file type
- how to sort files manually
- how to sort files in alphabetical order
- sort bam file
- how to sort your files
- samtools sort sam to bam
- how to view sam file
- how to sort file
- convert sam to bed
- how to sort files numerically in windows
- samtools index
Information related to the topic how to sort sam file
Here are the search results of the thread how to sort sam file from Bing. You can read more if you want.
You have just come across an article on the topic how to sort sam file. If you found this article useful, please share it. Thank you very much.